
Regulatory Services
MDAI provides regulatory consulting services for Medical Device companies required to comply with FDA regulations. We understand that becoming conversant with FDA regulations and successfully navigating the regulatory requirements can be very challenging. Whether your goal is to bring a new product to market or to modify an existing product line, Medical Device Approvals professionals have a thorough understanding of FDA Regulations and a proven track record of helping companies comply with the requirements. We will work with you from inception to approval, developing a comprehensive strategy to avoid delay and unnecessary expense while ensuring your FDA submission is cost-effective, timely and complete.
Regulatory Consulting Services include:
*Regulatory Strategy Development.
Medical devices are divided into three classes, each with different regulatory requirements based on risk stratification as outlined below.
Class |
Risk |
Regulatory Requirements |
Class I |
Present minimal potential harm to the user |
|
Class II |
More complex, higher risk that are not life-sustaining |
|
Class III |
Support or sustain human life |
|
Identifying the class and requirements, then putting together a complete regulatory strategy for the lifecycle of your device can be challenging. Some devices may be a Class III but can be cleared with a 510(k). Other devices may be a Class II but require clinical data.
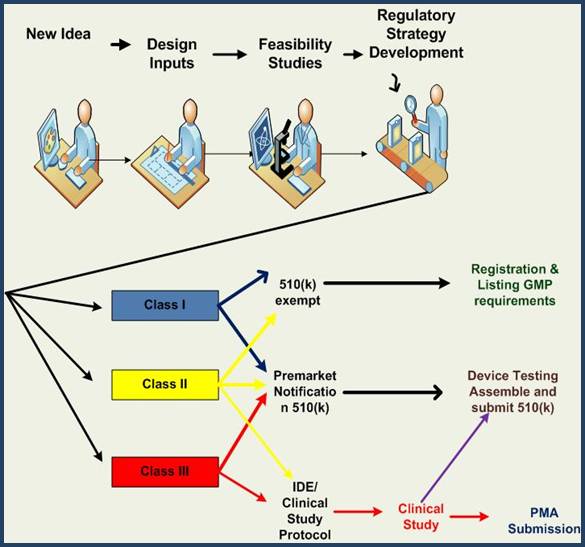
MDAI can provide your company with the following:
- Initial assessment to identify classification requirements, and standards for compliance with FDA guidance documents and how to show safety and efficacy of your device.
- Identification of predicate devices, if applicable, to ascertain regulatory pathways.
- Development of a comprehensive regulatory strategy to cover the entire product life cycle.
- Project management to supervise required testing and documentation.
Types of Marketing Applications :
Exempt Devices. These are low risk devices, usually Class I but some Class II devices also. They are exempted from premarket (510(k) or PMA) requirements and sometimes are exempt from GMP requirements. They do require an Establishment Registration, and a device listing. You cannot extend intended uses beyond what the regulation allows or you loose the exempt status and will need a 510(k) or PMA.
510(k)s. Some Class I devices require 510(k)s, most Class II devices require 510(k)s, and a few Class III devices require 510(k)s. You must be able to demonstrate that your device is substantially equivalent (SE) to some other device already on the US market, known as a predicate device. What this means is that your device is as safe as a very similar device already on the market in the USA. This route involves providing a very detailed comparison of the devices, assembling a file containing all the information, submitting it to the FDA or an acceptable third party for review and waiting for their go ahead before selling on the US market.
The 510(k) application will utilize design control principles (except in the case of exempt devices). There are three types of 510(k)s:
- Traditional Premarket Notification – Most companies introducing a new Class II medical device must submit a full 510(k).
- Abbreviated 510(k) submission is possible if FDA guidance documents exist, special controls have been established or you have followed a an FDA published consensus standard. For an abbreviated 510 (k) you provide summary reports that demonstrate your use of guidance documents, special controls or Declarations of Conformity to recognized standards to expedite review of your submission.
- SPECIAL 510(k) submission allows a manufacturer to modify their own device that already has 510(k) clearance. It allows the manufacturer to declare conformity with the Design Controls set out of 21 CFR Part 820 without providing the data. The special 510(k) also applies to Pre-amendment Devices (those approved before May 28, 1976).
Pre-Market Submissions (PMAs) A PMA submission is the most rigorous application and is used to demonstrate to the FDA that a new device or therapy is safe and effective.
- Many Class III devices are implants or provide life-sustaining therapies.They require PMAs as this is the highest risk classification in the US.
- A Class II device can also require a pre-market approval if the FDA determines that the new technology introduces safety issues that have not been adequately addressed by testing.
- Human use data from a formal clinical study is almost always required in addition to laboratory studies.
Investigational Device Exemption (IDEs) Acceptance by the FDA of an IDE application allows a sponsor to use an un-cleared or unapproved medical device in a clinical study to collect safety and effectiveness data to support a 510(k) or PMA submission. Sponsors must obtain IDE approval before the study begins. An approved IDE allows the device to be shipped lawfully for use in the study without complying with other FDA requirements that would apply to devices in commercial distribution. MDAI will work with you to plan, write, edit, organize and submit your IDE application, which typically includes:
- Detailed description of the device and an explanation of how it works
- Prior studies performed on the device
- Investigational Plan (protocol, case report forms, informed consent form, investigator agreement, risk analysis, statistical plan, and monitoring procedures),
- Labeling and device labels appropriate for the clinical study
- Summary of manufacturing procedures.
To discuss how Medical Device Approvals can help you achieve your regulatory objectives, please contact us at kathleen@mdapprovals.com